§1 Super-resolution optical imaging breakthrough in optical microscopy

Optical microscopes have long been an important tool in biomedical research due to their non-contact and non-destructive advantages. However, since 1873, it has been believed that the resolution limit of optical microscopes is approximately 200 nm and cannot be used to clearly observe biological structures with dimensions within 200 nm. Super-resolution Optical Microscopy is the most significant breakthrough in the field of optical microscopy this century. It breaks the resolution limit of optical microscopes. In other words, it surpasses the resolution limit of optical microscopes, so it is called super-resolution. Optical imaging provides unprecedented tools for life science research.
According to Abbe’s law, when a point light source passes through a perfect optical system, due to the diffraction effect of light, it will eventually form a blurry spot image (Airy disk), and the full width at half maximum of the spot satisfies FWHM≈0.61λ /NA, where λ is the operating wavelength of the optical system and NA is the numerical aperture of the optical system.
The above formula is also a description of the full width at half maximum of a point spread function (PSF). It shows that if the particles within this radius are illuminated at once, they cannot be distinguished using optical microscopy. Therefore, this formula is also a description of the resolution of optical microscopy.
The resolution of an optical imaging system depends on the minimum distance between two point light source images. If you want to improve the imaging resolution, you need to reduce its operating wavelength or increase the numerical aperture of the system (usually using a high numerical aperture objective lens).
For common optical microscopes working in the visible light band, the resolution is approximately 200nm. So how to further improve the imaging resolution of the system under the existing circumstances, so as to achieve resolution imaging that breaks through the diffraction limit?
With the continuous exploration of scientific researchers, resolutions less than 100nm have been achieved through a variety of different optical system designs. These high-resolution optical microscopy technologies that can break through the diffraction limit imaging are called super-resolution imaging technologies.
According to their different principles, existing super-resolution imaging technologies can be divided into two major categories:
- One type is based on single-molecule positioning imaging methods, which use the photoswitching characteristics of special fluorescent molecules or other mechanisms to randomly excite sparse fluorescent points at different times, and position the fluorescent molecules through corresponding algorithms;
- One type is to transform the point spread function of the imaging system to achieve imaging that breaks through the diffraction limit resolution.
Super-resolution optical imaging specifically refers to microscopes whose resolution breaks the resolution limit of optical microscopes (200nm). The technical principles mainly include stimulated emission loss microscopy technology and light-activated positioning microscopy technology.
Two technical principles of super-resolution optical imaging
Super-resolution optical imaging technology usually refers to super-resolution imaging technology based on far-field optical microscopy, which mainly includes two implementation methods: one is a super-resolution imaging method based on special intensity distribution illumination light field (such as STED). The other is methods based on single-molecule imaging and localization (such as PALM).
1. Stimulated Emission Depletion Microscopy (STED)
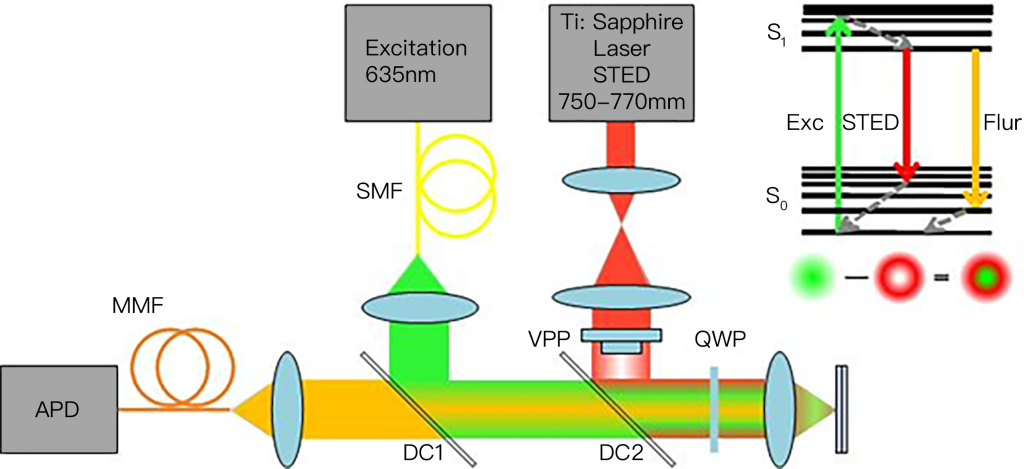
This imaging theory is derived from Einstein’s stimulated emission theory. German scientist Stefan W. Hell creatively used stimulated emission to suppress autofluorescence radiation, and finally realized stimulated emission depletion microscopy (STED). . A typical STED microscope requires two strictly coaxial laser beams, one of which is excitation light and the other is loss light (also called STED light). Fluorescent molecules within the Airy disk are excited using excitation light, and their electrons transition from the ground state to the excited state. Subsequently, a donut-shaped (similar to a lifebuoy shape) loss light is used to irradiate the sample, causing the excited molecules at the periphery of the excitation spot to release energy and return to the ground state by stimulated radiation, while the excitation molecules located in the inner region of the excitation spot are The molecules in this state are not affected by the loss of light and continue to return to the ground state by autofluorescence. This combination of illumination methods limits the fluorescence emission area to an area smaller than the Airy disk, obtaining a fluorescence luminescence point smaller than the diffraction limit. Finally, by scanning coaxial excitation light and loss light in two-dimensional (or three-dimensional) space, a two-dimensional (or three-dimensional) super-resolution image is obtained. In 1994, Stephen Hull and others proposed the theory of STED microscopy. In 2000, Stephen Hull’s research group confirmed the super-resolution imaging capability of the STED microscope through biological experiments.
2. Photoactivation Localization Microscopy (PALM)
The Abbe limit states that the image formed by two fluorescent molecules separated by 1/2NA cannot be resolved in the far field, but there is no limit on the accuracy of determining the center position of a single fluorescent molecule. If only one molecule emits fluorescence in the Airy disk, we can use a single-molecule positioning algorithm combined with the shape of the Airy disk of the optical system to obtain the center position of the fluorescent molecule with ultra-high precision (nanometer scale). If this single-molecule positioning idea is used to achieve super-resolution imaging, the key lies in how to distinguish multiple fluorescent molecules within an Airy disk.
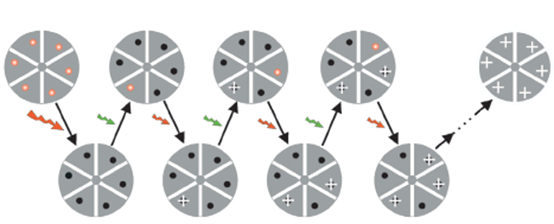
In order to overcome the limitation that only one molecule is allowed to emit fluorescence in an Airy disk, in 1995, American scientist Eric Betzig proposed through theoretical analysis that spectral characteristics can be used to control the emission wavelengths of different molecules in the Airy disk. Fluorescent molecules perform time-sharing detection and central position positioning, thereby achieving super-resolution imaging of fluorescently densely labeled samples. In 2006, Eric Betzig and others used the controllable fluorescence switching characteristics of light-activated green fluorescent protein (PA-GFP), combined with a single-molecule positioning algorithm, to achieve super-resolution imaging of biological samples. They first used a low-energy 405 nm laser (activation light) to sparsely activate PA-GFP, and then used a 561 nm laser (excitation light) to perform single-molecule fluorescence imaging of the activated PA-GFP until the activated PA-GFP molecules Photobleached. By repeating the activation-excitation-localization-bleaching process, the center positions of a large number of PA-GFP molecules can be found with high precision within the Airy disk, thereby reconstructing a super-resolution image consisting of the center positions of PA-GFP molecules. This technology is called Photoactivation Localization Microscopy (PALM).
In 2006, Xiaowei Zhuang’s group at Harvard University proposed Stochastic Optical Reconstruction Microscopy (STORM). Its imaging principle is similar to PALM, but the scheme to achieve rarefaction of fluorescence emission in densely labeled samples is different.
§2. Super-resolution imaging based on single-molecule localization
The first type of method is based on super-resolution imaging achieved by molecular localization, mainly including photoactivated localization microscopy (PALM) and stochastic optical reconstruction microscopy (STORM).
Fluorescent proteins with photoswitchable properties are key to this type of technology. In 2002, Patterson et al. first discovered a fluorescent protein, PA-GFP, which does not emit light in an unactivated state. After being activated with a 405nm laser for a period of time, it can be excited by the 488nm excitation light and emit a green fluorescence signal. In 2006, Betzig et al. used this fluorescent protein with switching properties combined with single-molecule fluorescence imaging technology to first propose PALM.
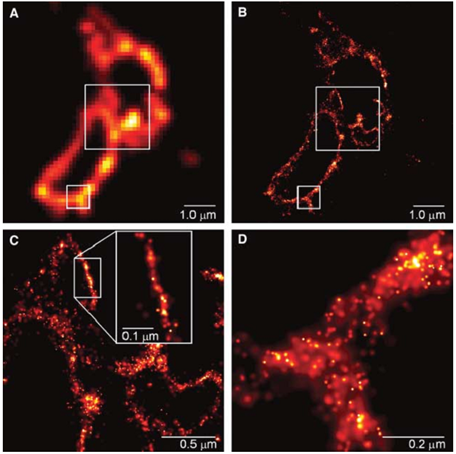
Taking advantage of the switching characteristics of the fluorescent protein Cy5 under different excitation wavelengths, “Stochastic Optical Reconstruction Microscopy (STORM)” was proposed.
When two luminophores are too close, conventional microscopes cannot resolve them. Zhuang Xiaowei’s idea is that if two colors of light can be used, one beam can overexcite most particles to the dark state through photobleaching; the other beam can reactivate a very few dark state particles through activation. Because very few are activated. In this way, there are some particles that only emit light by themselves and the surrounding areas do not emit light. You can easily locate them, excite them, bleach them, and then excite other particles, and so on.
Moreover, she discovered an important Cy3-Cy5 pair that emits light of different colors. This pair of fluorophores was used in fluorescence resonance energy transfer FRET imaging studies in the past. Zhuang Xiaowei accidentally discovered during his research on cold viruses that this pair of cute proteins can be like a switch, controlling them to fluoresce or not to fluoresce through light.
The principles of PALM and STORM are roughly the same. They both use repeated activation and bleaching of fluorescent molecules to obtain sparse position information of fluorescent molecules. After hundreds of iterations, the precise positioning of all fluorescent molecules in the cell is reconstructed to On the same image, a microscopic image with a resolution at least 10 times higher than that of a traditional optical microscope can finally be obtained.
The difference is that STORM can be used to study the super-resolution localization of endogenous proteins in cells. However, it can be found from the basic principles of PALM and STORM that these two types of super-resolution imaging technologies still rely on traditional optical microscopy systems, and their optical point spread functions have not changed, that is, the single-shot imaging resolution is still at the diffraction level. In the limit, it is “trading time for space”, obtaining high spatial resolution by sacrificing temporal resolution.
§3. Super-resolution imaging based on point spread function transformation
This type of method compresses the point spread function (PSF) of the optical system by modifying the light source to achieve super-resolution imaging. This type of imaging method mainly includes stimulated emission depletion (STED) microscopy technology and structured illumination microscopy technology. (structured illumination microscopy, SIM), reversible saturable optical linear fluorescence transitions (reversible saturable optical linear fluorescence transitions) microscopy technology, etc.
STED microscopy technology was first proposed in 1994 by Stefan W. Hell et al. It uses stimulated emission loss to change the point spread function of fluorescence emitted by fluorophores. Some fluorescent molecules are in an excited state under excitation light illumination, but when irradiated with another laser beam with a longer wavelength than the excitation light, the fluorescent molecules can be quenched from the excited state back to the ground state, that is, they emit light when two laser beams are irradiated at the same time. . Based on such a fluorescence characteristic, Hell et al. used one beam of laser to irradiate the sample and at the same time used another beam of high-energy pulse laser to form a close ring laser illumination. The wavelength was slightly longer than the excitation light. In this way, most of the fluorescence excited by the short wave passed through the receptor. The process of stimulated emission loss is quenched, compressing the point spread function and improving the resolution of the microscope. However, STED imaging requires point scanning of the sample, and the laser light intensity used is too high, which may cause photobleaching or even phototoxicity of the observed sample. At the same time, the optical path of STED imaging is complex and the system construction cost is also high.
In Figure 3, the green arrow represents that the particle is excited from the low energy level S0 to the high energy level S1, and then the particle relaxes to the metastable state – the lowest point of the high energy level. Next, the particle will stay here for a short time (as short as a few ns), and then the particle will return to the low energy level S0. This is the choice for most electronics. When jumping down, they will land at different heights of the S0 energy level and form a certain distribution.
Still in Figure 3, if the minimum PSF of the fluorescence phenomenon caused by the green arrow is the radius of the green circle, if you put a red eraser on it (the wavelength of the stimulated radiation of the particles is the same), then there will be a few remaining There are more and more fluorescences in the middle, which reduces the point spread function and achieves super-resolution.
In terms of experimental operation, select a suitable fluorescent substance, give it an excitation pulse (about 2ps) in sequence, and then give it a stimulated emission wavelength pulse (about 250ps) as soon as it transitions up, and then use a dichroic mirror to distinguish Stimulated emission and spontaneous emission, detected spontaneous emission signals. The larger the stimulated emission (the eraser is clean), the smaller the remaining PSF is, which means the higher the resolution. This is Pulsed STED. It can also give continuous signals, because dichroic mirrors can distinguish them, but they are not so clean. This is CW STED.
Unlike STED, which requires point-by-point scanning imaging, SIM is a wide-field microscopy imaging technology that can achieve higher imaging speeds and is more suitable for biological research in vivo and with low bleaching. SIM uses non-uniform sinusoidal stripe illumination light to illuminate the sample. When the fine topography of the measured sample and the stripe-like illumination light are superimposed on each other, a “Moiré fringe” with a lower spatial frequency than both can be obtained. This moiré fringe contains information about both the sample and the illumination light. In other words, structured light illumination can be used to encode high-frequency sample information that cannot be collected by the optical system into moiré with a relatively low spatial frequency that can be collected. Stripe signal. Using post-image reconstruction technology, the high-frequency information of the sample can be reproduced on the obtained image, thus achieving super-resolution imaging that breaks through the diffraction limit. At the same time, using the method of multi-beam light interference, two-dimensional structured light illumination imaging can be extended to the field of three-dimensional imaging. At present, resolutions of about 100nm in the lateral direction and 200nm in the axial direction have been achieved. Although the resolution of SIM is lower than other super-resolution imaging technologies, its method is simple, does not require special fluorescent labels, and can achieve fast, dynamic, three-dimensional super-resolution imaging.
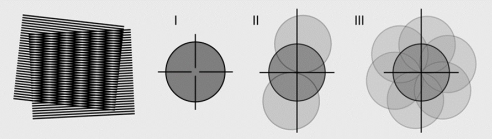
Confocal uses a point illumination and adds a pinhole to improve resolution. To be precise, since optical noise always causes interference in terms of resolution, using a pinhole to remove the optical noise can improve the resolution. Confocal can increase the resolution by 1.4 times, because even if the pinhole is as small as one point, according to the principle of reversible optical path, this point will be as large as a spread function when it reaches the sample. Ultimately, the point spread function of confocal is the point spread of the excitation light. Multiply the pinhole point expansion.
Confocal improves resolution by blocking reception, while SIM improves resolution by modulating the illumination light. If the structural modulation of SIM is placed at the receiving end through conjugation, SIM is also a one-dimensional modulation. Furthermore, by performing one-dimensional constraints from multiple angles, two-dimensional resolution improvements can ultimately be achieved. Currently, the more popular method is to do it every 120 degrees, and SIM can increase the resolution by 2 times.
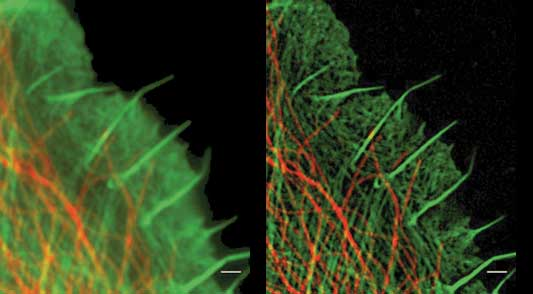
In general, due to the limitation of imaging resolution caused by optical diffraction effects, it is currently mainstream to sacrifice a certain time resolution to obtain imaging that breaks through the diffraction limit by positioning single molecules and modifying the point spread function of the optical system. method.
In addition to the far-field super-resolution imaging technology based on the principle of fluorescence imaging mentioned above, imaging that breaks through the diffraction limit can also be obtained using near-field scanning optical microscopy (NSOM). Near-field optical imaging is different from classical optics in that the probe is only a wavelength of the order away from the sample. It can collect near-field information that cannot be collected by far-field imaging, thereby obtaining imaging resolution comparable to that of an atomic force microscope.
§4. Research progress on super-resolution imaging technology in China
At present, extensive and in-depth research on super-resolution imaging technology has been carried out in China.
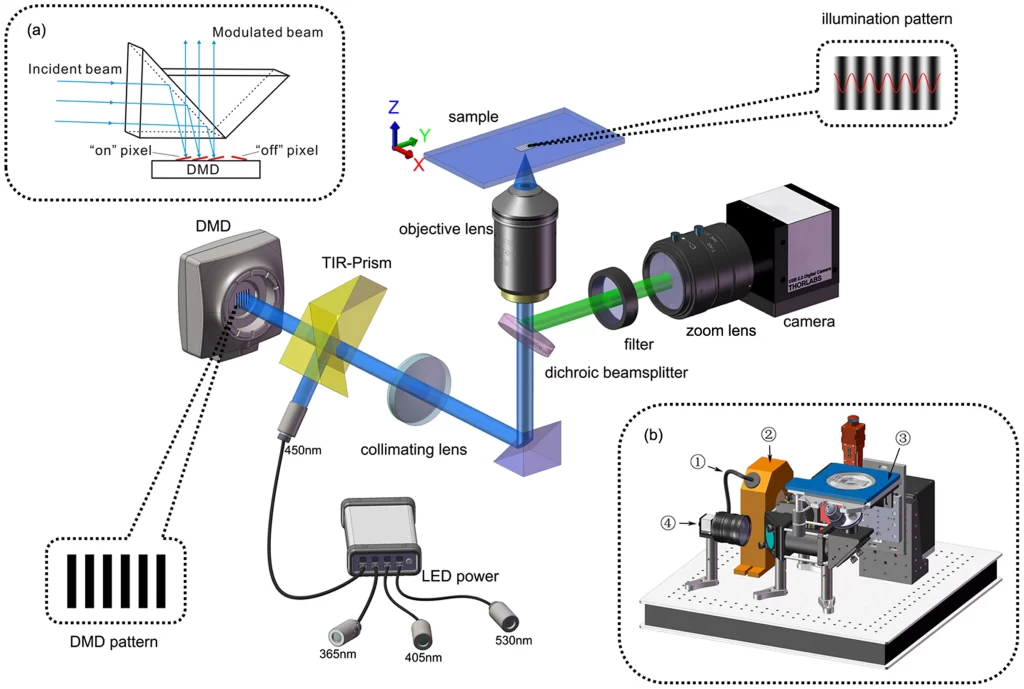
The Xi’an Institute of Optics and Precision Mechanics, Chinese Academy of Sciences, adopts a structured light illumination solution using digital micromirror devices (DMD) and LED lighting, which reduces background noise caused by unfavorable factors such as speckle interference and can achieve a spatial resolution of 90nm and a slicing speed of 190ms. .
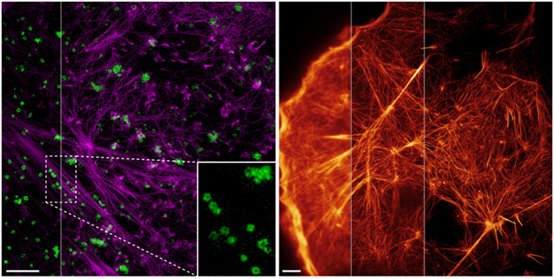
Left: Total internal reflection structured light microscopy imaging technology (highNA TIRF-SIM) with a two-color high numerical aperture objective. Purple: actin; green: endocytic vesicles (CCP). Right: Nonlinear structured light illumination (PA NL-SIM) imaging of agonist proteins tagged with nonlinear activation based on Skylan-NS.
In 2015, scientists from the Institute of Biophysics of the Chinese Academy of Sciences and the Howard Hughes Medical Institute of the United States published in Science that they used a new type of repeatedly activated fluorescent protein Skylan-NS and structured light-activated nonlinear SIM technology to obtain The image of the disassembly and self-assembly process of skeleton proteins during cell movement and shape change, as well as the dynamic process of tiny endocytic bodies called “caveolae” on the surface of the cell membrane, can reach a resolution of 62nm.
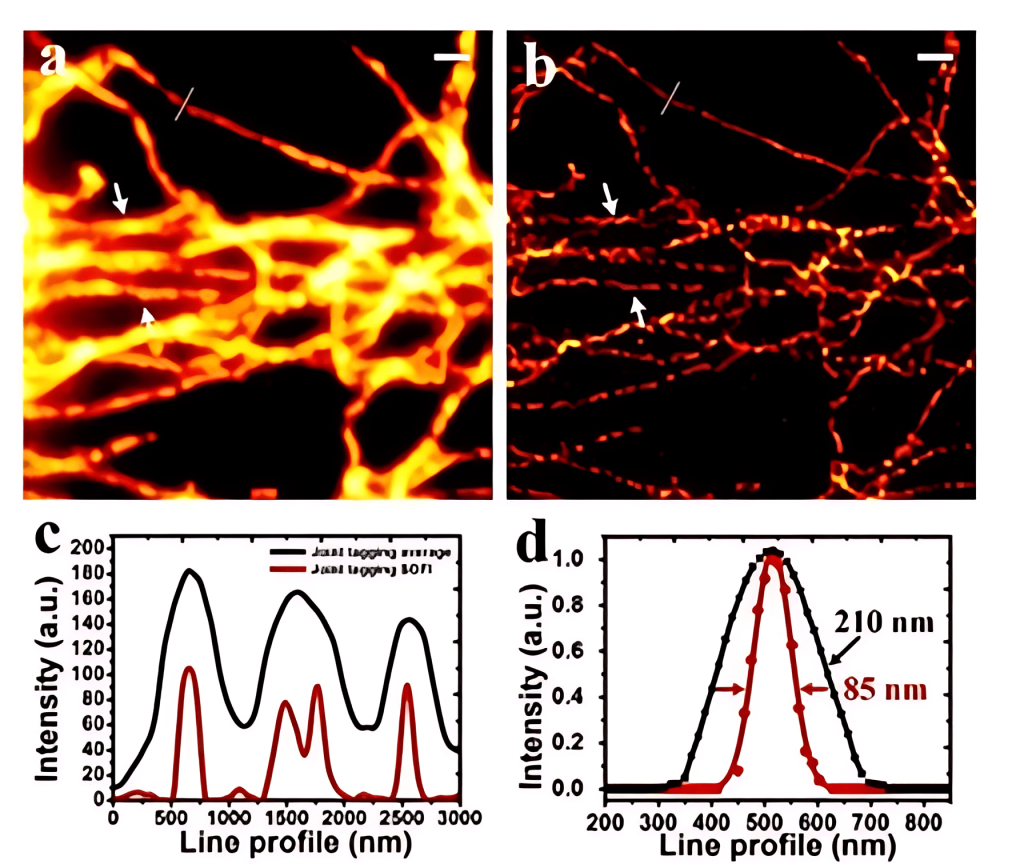
In 2015, Xi Peng from Peking University combined quantum dots, spectral methods and SOFI super-resolution technology to achieve ultra-high spatial and temporal resolution imaging of 85nm in 3 seconds on an ordinary wide-field microscope, using the combination of quantum dots with different emission wavelengths. Marking effectively reduces the artifacts of high-order imaging and more realistically restores the complete structure and detailed information of biological samples.
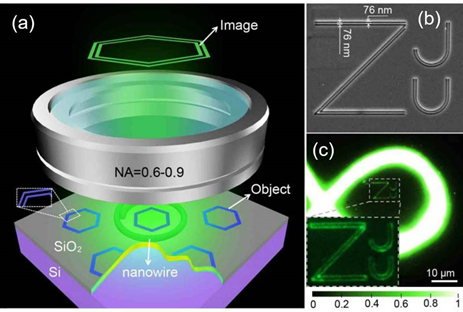
In 2017, Yang Qing and Liu Xu of Zhejiang University used nanowires as local light sources and proposed a technology called “Nanowire ring illumination microscopy (NWRIM)”, which achieved a large field of view for the first time. , label-free super-resolution imaging, the field of view currently reaches thousands of square micrometers, which is more than one order of magnitude larger than previously reported label-free far-field super-resolution microscopy methods.