——Breaking the shackles of Abbe’s diffraction limit
§1 What is the Abbe diffraction limit?
The resolution of optical microscopy is physically limited. This fundamental limit was first described by Ernst Karl Abbe in 1873, and although no equation is mentioned in this article, Abbe reported that the minimum resolvable distance between two points using conventional microscopy may never be less than half the wavelength of the imaging light .
In some of his later papers, he concluded that, due to diffraction, imaging resolution is limited to half the wavelength λ modified by the refractive index n of the medium and the angle θ of the focused light cone:
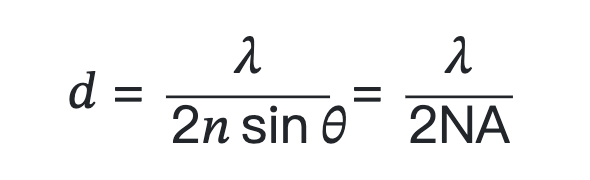
Numerical aperture (NA) and resolution limits are shown in Figure 1.
The formal expressions for lateral and axial resolution in optical microscopy proposed by Abbe are:
Resolution x, y = λ/2[ηsin(α)]
Resolution z = 2λ/[ηsin(α)]2
where λ is the wavelength of light (excitation in fluorescence), eta represents the refractive index of the imaging medium, and the combined term eta sin(α) is called the objective numerical aperture (NA).
The limit is basically a result of the diffraction process and the wave nature of light. The high-frequency components that give images their clarity are lost due to the finite numerical aperture of the lenses that collect the light. This results in a blurry appearance of the captured image. In a more mathematical sense, it can also be said that the resulting image is the convolution of the actual object with the so-called point spread function (PSF) of the optical system. PSF is the response of an optical system to a point emitter due to the diffraction limit and imperfections in the optical system. In a perfect optical system without any aberrations, the PSF can be well described by the so-called Airy function. Due to the diffraction limit, if the distance between two emission points is smaller than the diffraction limit, they cannot be optically resolved, as shown in Figure 1(b). This definition of microscopy resolution is also often referred to as the Rayleigh criterion.
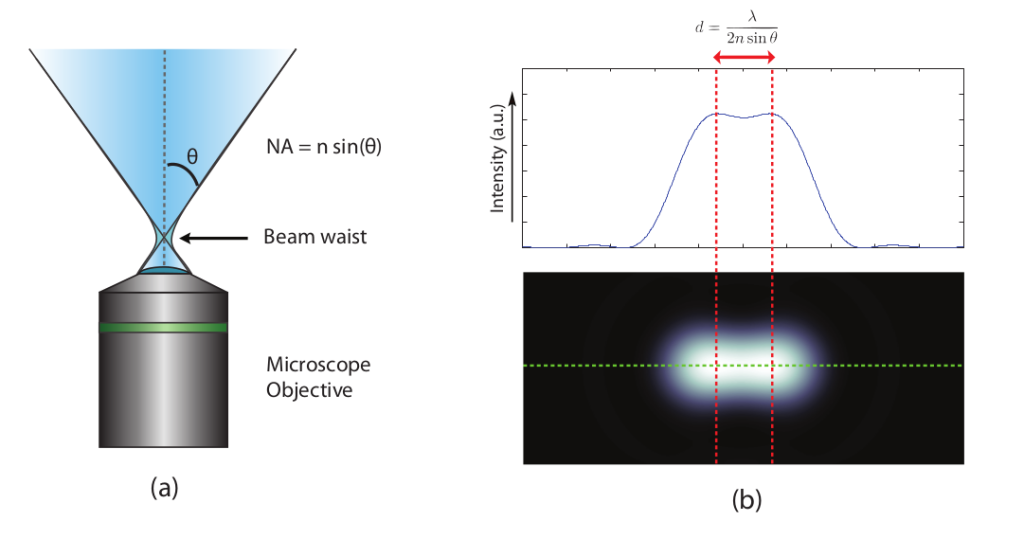
The diffraction limit was considered unbreakable for a long time. This was before new state-of-the-art optical super-resolution techniques were developed over the past two decades. STED is one of the super-resolution techniques that uses modern technology to circumvent the diffraction limit. Some other super-resolution technologies include STORM, PALM, and GSD.
§2 Optical Microscopy Revolution: Super-resolution Microscopy

In the early 1990s, instruments with opposed objectives (technically designated 4Pi and I5M) developed by Stefan Hell, Mats Gustafsson, David Agard, and John Sedat were able to reduce axial resolution using confocal and widefield configurations, respectively. Increase to about 100nm. However, even though these instruments were able to improve axial resolution by a factor of five, lateral resolution was still not improved. In the late 1990s, new microscopy techniques pioneered by Stefan Hell were able to overcome the diffraction limitations of Abbe’s lateral resolution, which ultimately led to a revolution in fluorescence microscopy.
This new method, now collectively known as super-resolution microscopy, is characterized by both lateral and axial resolutions of tens of nanometers or less. What all these new technologies have in common is their ability to resolve features below the diffraction limit by turning fluorophores on and off in a temporal sequence, allowing for continuous recording of the signal.
The most important advances in super-resolution imaging have been achieved in so-called far-field microscopy, involving spatially or temporally modulated transitions between two molecular states of a fluorophore (such as switching between dark and bright states), or By physically reducing the size of the point spread function (PSF) used in excitation illumination.
Among the methods of improving resolution by modifying the point spread function (PSF), the most important techniques are known by the abbreviations STED (Stimulated Emission Depletion; from the laboratory of Stefan Hell) and SSIM ((Saturated) Structured Illumination Microscopy) ; Pioneered by Mats Gustafsson). Techniques that rely on the detection and precise localization of single molecules include PALM (photoactivated localization microscopy) and STORM (stochastic optical reconstruction microscopy).
There are many variations of these techniques, as well as advanced methods that can be combined with or even improve the performance of existing imaging solutions. What’s more, relative to advances in traditional microscopy techniques, new super-resolution techniques are being introduced at an almost breathtaking pace, and it is believed that at some point in the near future, single-nanometer resolution is likely to be achieved. Implemented in commercial instruments, this is not unreasonable.
| Super-resolution microscopy imaging technology | principle | Live cell imaging | XY resolution | Z resolution | Fluorophore |
| STED | stimulated emission of radiation | Yes | ~35nm | ~500nm | any |
| isoSTED | 4Pi Optical | Yes | ~30nm | ~30nm | |
| RESOLFT | reversible saturation | Yes | ~35nm | ~500nm | Light-controlled reversible transformation |
| 4Pi RESOLFT | 4Pi Optical | Yes | ~30nm | ~30nm | |
| SIM | structured light illumination | Yes | ~100 nm | ~500nm | any |
| NL-SIM | Nonlinear Optics | Yes | ~60 nm | ~500nm | |
| STORM | single molecule localization, reconstruction | Yes,fast detector | ~25nm | ~50nm | photo-controlled switchable |
| Dual objective lens, PSF modification | ~10nm | ~30nm | |||
| PALM | photosensitive fluorescence single molecule localization | Yes,fast detector | ~25nm | ~50nm | Light-controlled reversible transformation |
| iPALM | dual objective lens, fluorescence interference | ~25nm | ~10nm | ||
| DNA-PAINTDMI | single molecule localization, point accumulation | NO | ~5nm | ~50nm | DNA-Dye |
| MINFLUX | integrated STED and PALM | Yes | 1nm | 1nm | any |
| ExM | physical expansion sample | NO | 25nm | 50nm | any |
Table 1 Technical characteristics of various types of super-resolution microscopy
§3 Stimulated Emission Depletion (STED)
STED super-resolution microscopy was developed based on a theoretical model proposed in 1994. This technology was first implemented in 1999 and used in materials research, and was subsequently applied in the field of life sciences in 2000. Since then, STED has revolutionized the fluorescence imaging paradigm. STED is the first realized super-resolution microscopy based on the target switching principle.
Stimulated radiation depletion microscopy (STED) is a fluorescence microscopy technique that overcomes the diffraction resolution limitations of confocal microscopy. The resolution is improved primarily by switching off the fluorescence of the dye molecules using intense laser stimulation of emission in regions outside the diffraction-limited excitation focus. This intense radiation causes nearly all excited molecules to return to their ground state, and then the fluorescence of the remaining excited dye molecules at the center of the excitation focus is detected and used to form high-resolution images.
The resolution can be further improved by performing time switching (gated STED, gSTED) processing on the collected data. Since STED can effectively narrow the observation range, in this case, collecting data at different observation volume diameters can help to clearly see the complex two-dimensional diffusion conditions in heterogeneous samples such as biofilms.

The STED method uses pairs of synchronized laser pulses. The first laser pulse is generated by, for example, a picosecond pulsed diode laser and is used to excite the fluorescent dye and produce a common diffraction-limited focus. The excitation pulse is followed by a depletion pulse whose frequency coincides with the emission spectrum of the dye. By spatially arranging the STED pulses into a donut shape using specially designed phase plates, only fluorescence from molecules peripheral to the excitation focus is quenched by stimulated emission. At the center of the donut, the STED laser intensity is zero and fluorescence is unaffected and detected by a single-photon sensitive detector.
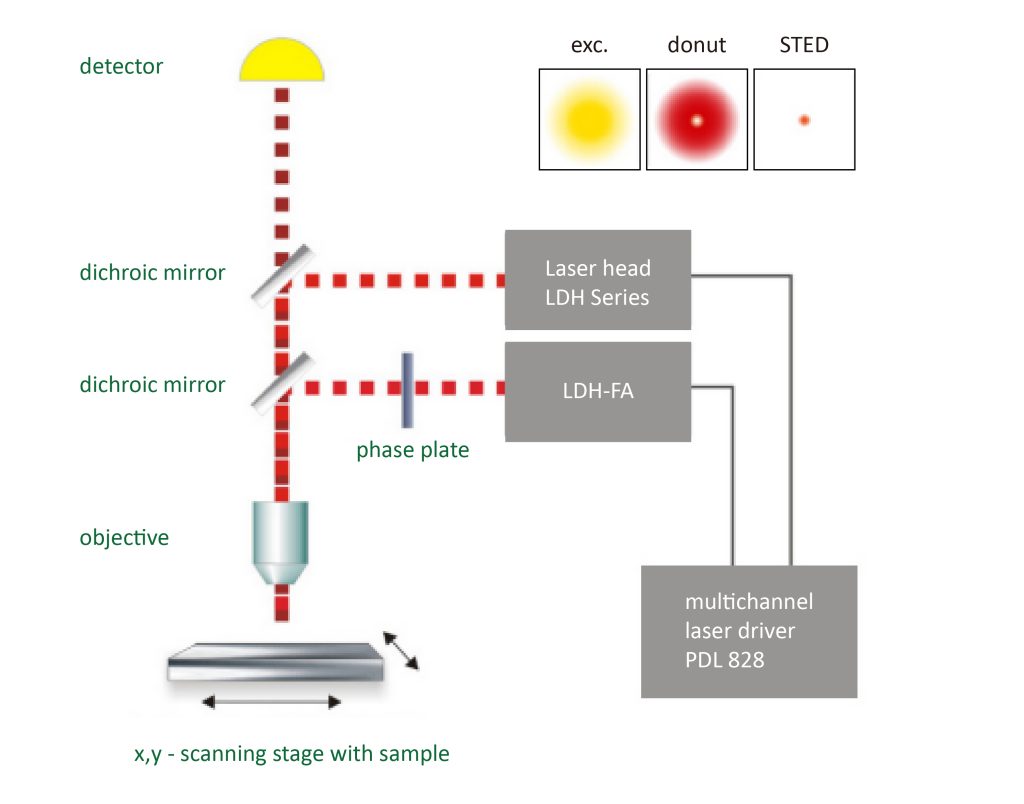
§4 4Pi fluorescence super-resolution microscopy combined with STED technology
Since most biological structures have a three-dimensional spatial distribution, extending super-resolution imaging to the three-dimensional level can clearly display these biological structures or record molecular motions. There are many technical ways to extend the original two-dimensional super-resolution microscopy imaging to the three-dimensional level.
For single molecule localization microscopy (SMLM), such as photoactivated localization microscopy (PALM), stochastic optical reconstruction microscopy, and fluorescence activation localization microscopy (fPALM), bifocal plane imaging or point imaging can be used Diffusion function (PSF) control method; for target switching super-resolution microscopy represented by stimulated emission depletion microscopy (STED) and reversible saturation optical fluorescence transition microscopy, the most commonly used strategy is to simultaneously Use two phase plates to create a three-dimensional hollow STED/RESOLFT focused spot through incoherent superposition.
Regardless of which technique is used, however, the ultimate resolution still depends on the sharpness of the microscope’s focused spot. A consequence of this fact is that it is relatively more difficult to increase axial resolution than to increase lateral (in-focal plane) resolution. Therefore, even though the lateral resolution can be increased to 20~40nm through super-resolution microscopy, the axial resolution achieved under the same architecture is still lower than the lateral resolution (the latter is about 2.5 times the former).
For optical mirrors, a single lens can only cover half of the spherical wavefront. Unless lateral resolution is deliberately sacrificed, it is very difficult to achieve three-dimensional isotropy. If there is an optical system that can collect the other half of the spherical wavefront behind the focal plane, the shape of the system PSF can be maintained (essentially) as a sphere due to the restoration of symmetry. Based on the above ideas, in the early 1990s, Hell et al. invented a 4Pi microscope based on two opposing objectives. In practical applications, one objective lens can only cover a semi-aperture angle of approximately 65°, while using two objective lenses can almost completely cover the entire solid angle of 4 π (actually it should be 2.5 π~3 π).
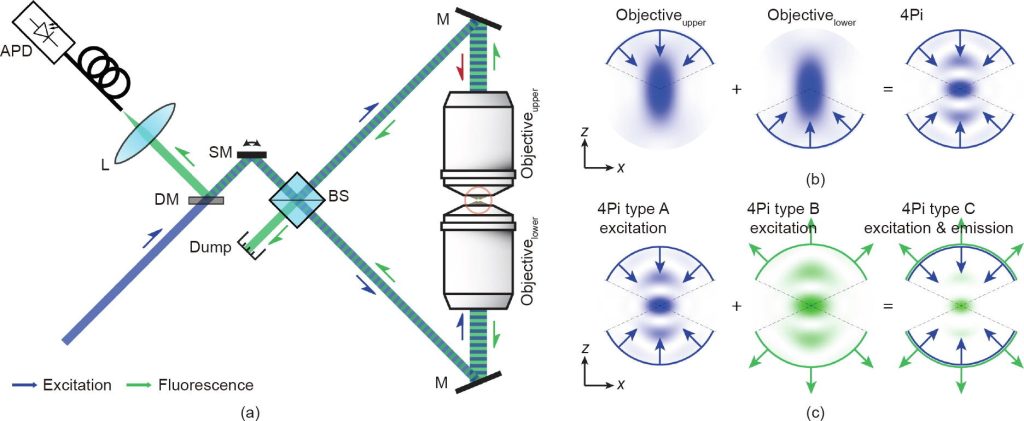
The combination of 4Pi microscopy and super-resolution microscopy can further improve the three-dimensional resolution of the system, especially the axial resolution.
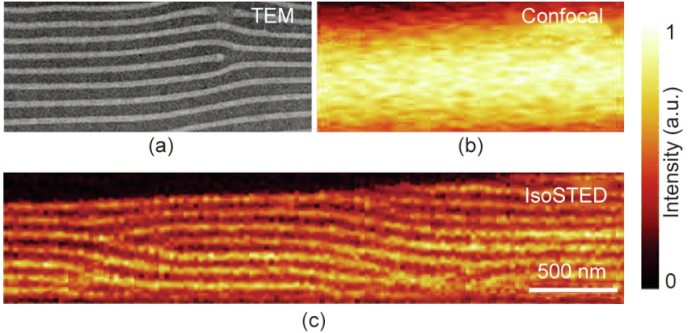
Technological advances over the past 15 years have brought STED microscopy into a new era of three-dimensional imaging. Among the many specific implementation methods, the 4Pi architecture based on two opposing objective lenses is particularly eye-catching because it can generate very sharp STED focused dark spots in the axial direction. Although its resolution advantage has not been fully utilized, the early so-called STED-4Pi microscopy has been able to provide an axial resolution of approximately 33nm near the focal plane. As an improved version of isoSTED microscopy, it can already provide isotropic three-dimensional high-resolution images.
§5 Development and future of super-resolution microscopy
Super-resolution fluorescence microscopy has transformed our understanding of the structure and function of many biological systems. However, challenges remain and further technological advances are required to maximize the impact of super-resolution microscopy.
The spatial resolution achieved by super-resolution microscopy in biological systems is typically between 10nm and 70nm, which is larger than most biological molecules. Achieving true molecular-scale resolution (approximately 1 nm) allows direct probing of molecular interactions and conformations within cells, but remains a challenging task.
In principle, the two major types of optical methods that overcome the diffraction limit, including pattern illumination methods represented by STED, RESOLFT and NL-SIM, and single-molecule switching methods represented by STORM, PALM and PAINT, can achieve infinitely high resolution. Rate. However, some practical factors, such as the requirement to increase illumination intensity (the former) and increase the phosphor photon budget (the latter) to obtain higher resolution, limit the achievable resolution.
In addition, although super-resolution imaging has sub-second or even millisecond temporal resolution in some cases, due to the trade-off between spatial and temporal resolution, the limited photon budget of fluorophores, and phototoxicity to the sample, Live cell imaging with high spatiotemporal resolution over long periods of time remains difficult and is an area of active development. In addition, in vivo super-resolution imaging deep inside tissues remains challenging despite extensive work on eliminating tissue-induced background, aberrations, and light scattering.
Another challenge, and an exciting new direction, is to increase the number of molecular species that can be imaged simultaneously. Cells contain thousands of different genes and other molecules that work together to produce behavior and function, but multicolor imaging can typically only observe a few different molecular species simultaneously.
Recent advances have pushed new boundaries in this direction, with genome-scale imaging now within reach. For example, single-cell transcriptome imaging methods can simultaneously image the RNA of 1,000 or more genes in a single cell by using multiplex fluorescence in situ hybridization (FISH) or in situ sequencing. In the future, DNA and proteins may reach similar levels of multiplicity. Combining these methods with super-resolution microscopy makes it possible to achieve genome-scale super-resolution imaging. Technically, a major challenge in genome-scale imaging is molecular crowding, which may hinder the resolution of adjacent molecules by conventional imaging, and super-resolution microscopy offers a promising solution.
From a biological perspective, the ability to image all molecules in complex molecular mechanisms or entire signaling pathways, and ultimately at the genome-wide scale, will provide a comprehensive picture of the molecular basis of cellular behavior and function. Excitingly, imagining a picture of the cell – with all its molecules imaged at a resolution that allows direct inference of molecular interactions – will lead to new opportunities for understanding life at the molecular level.